Samples & treatments
Nice neat metadata for analysis
Before we go any further, it is worth making sure we have all the sample and treatment information we need to make our analysis repeatable (by ourselves or someone else e.g. a reviewer).
More metadata is required for submitting to a repository but we will cover that later. For now, this is what you will need to perform an untargeted analysis.
You need to create two .csv files (you can do this in excel, R, google sheets, whatever you like) as long as the order and headings of the columns are as follows:
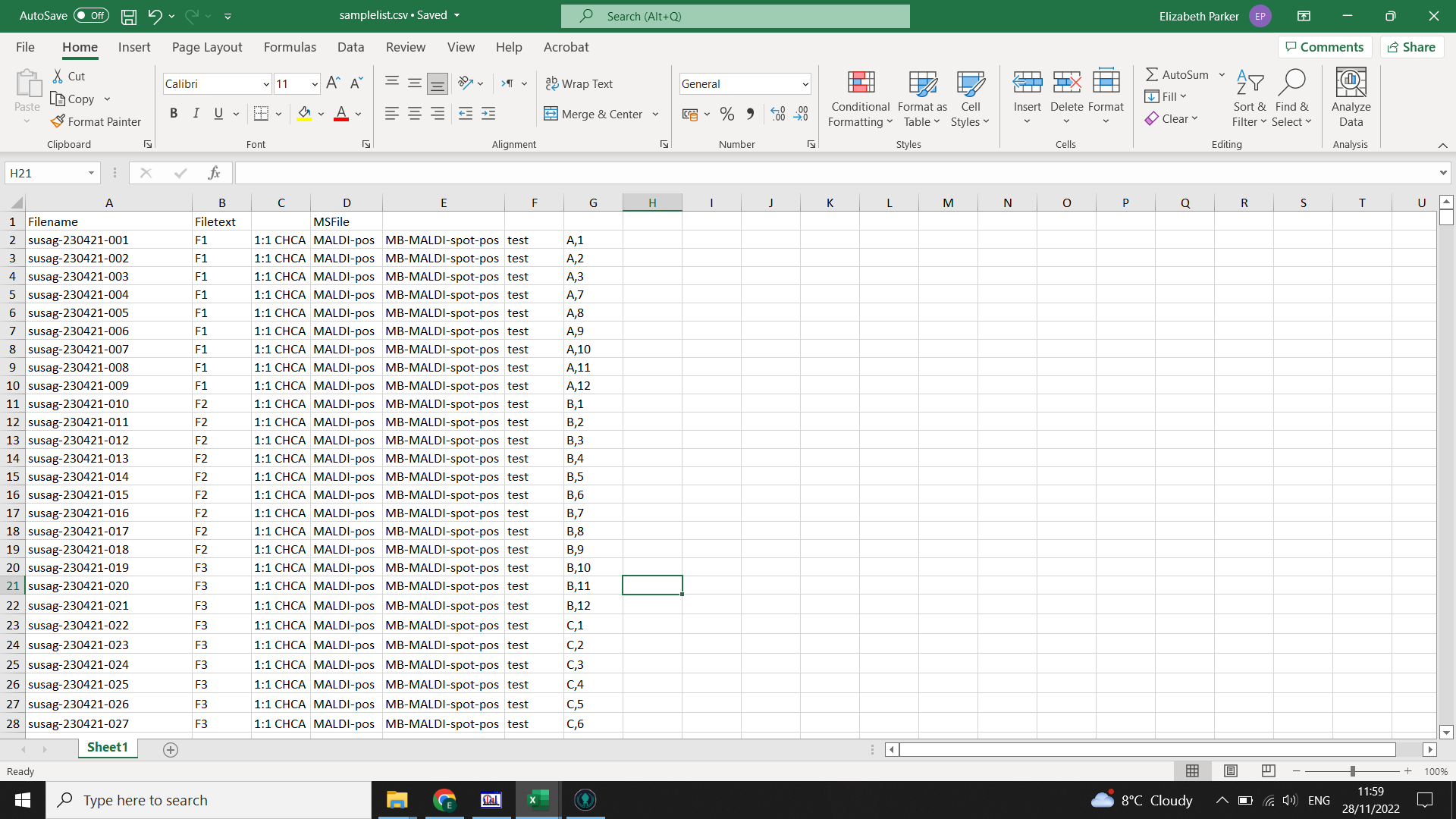
For you samplelist.csv you need the following columns which can be obtained from your MassLynx Sample List:
- “Filename” : this is a list of the filenames of your mzml files (the part before the
.mzml) - “Filetext” : this is the name you have manually added to the metadata of that sample in MassLynx1
- “MSFile” or an equivalent column that contains either “pos” or “neg” within it Any other columns will be ignored in this file
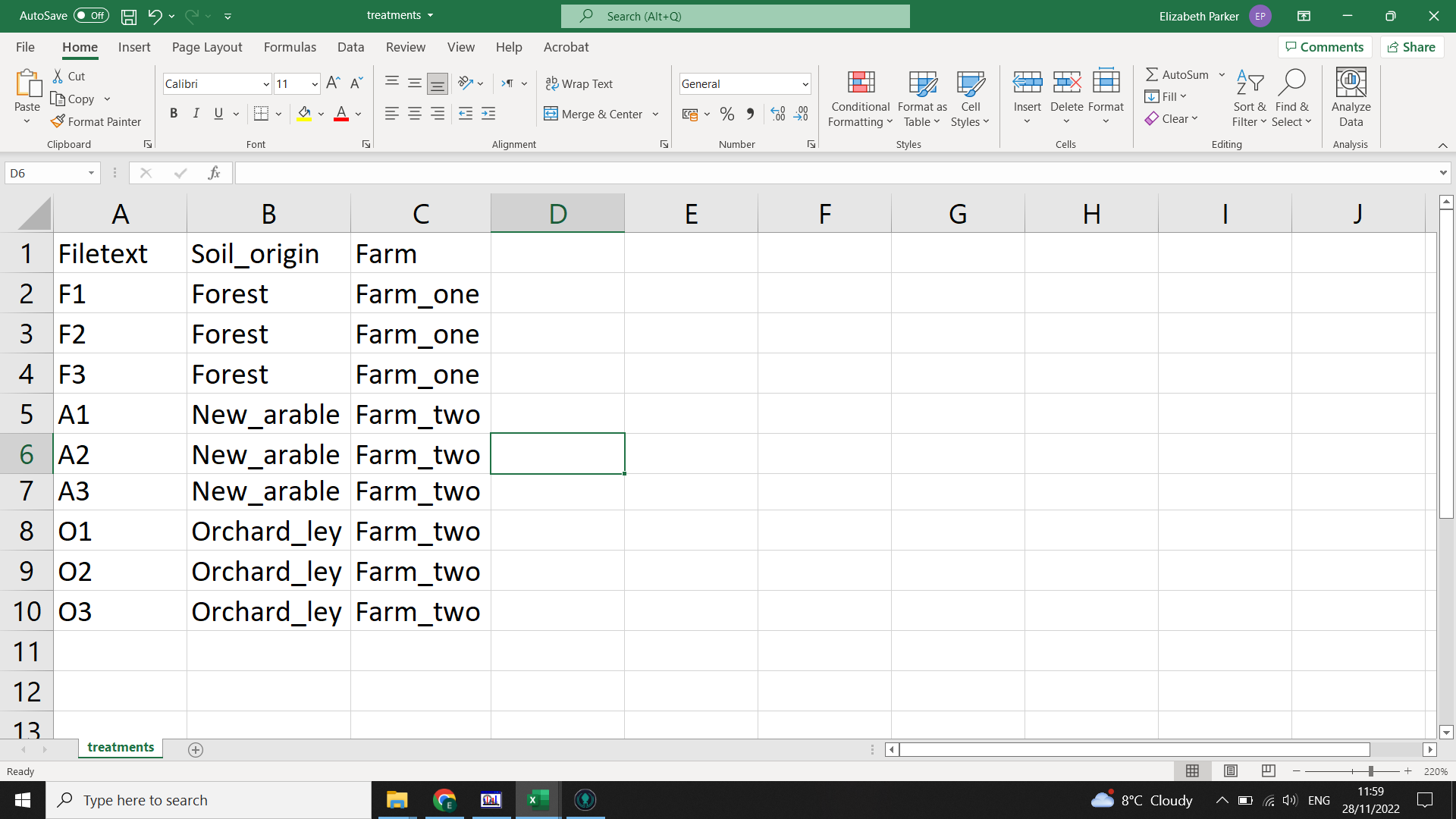
For your treatments.csv you need at least two columns (but can have as many as you like):
- “Filetext” : this must contain all the distinct values of Filetext from samplelist.csv
- “Variable1” : you can call this column whatever you like (but avoid spaces, instead use “-” or “_"). If you have compared a wild-type to a control for example, you might call this column “treatment” and fill it with “WT” and “C”
- “Variable2” etc : further variables that you wish to analyse. This may include batch identifiers (for example if you have many samples and they were run over multiple days), treatments or environmental variables
Keep these in a file with your .mzml data
-
You can find this at
$$ SampleID:in the_HEADER.txtfile of the original.RAWfolder if you need to ↩︎